Trim Galore Not Removing 0 Length Reads
Pre-processing of sequencing reads
The raw information obtained from the sequencing machine need to exist pre-candy.
Pre-processing can include the quality control of initial reads and read trimming that includes removing adapter sequences, filtering out low quality reads and trimming reads off depression quality base of operations pairs.
Quality control of sequencing reads
FastQC
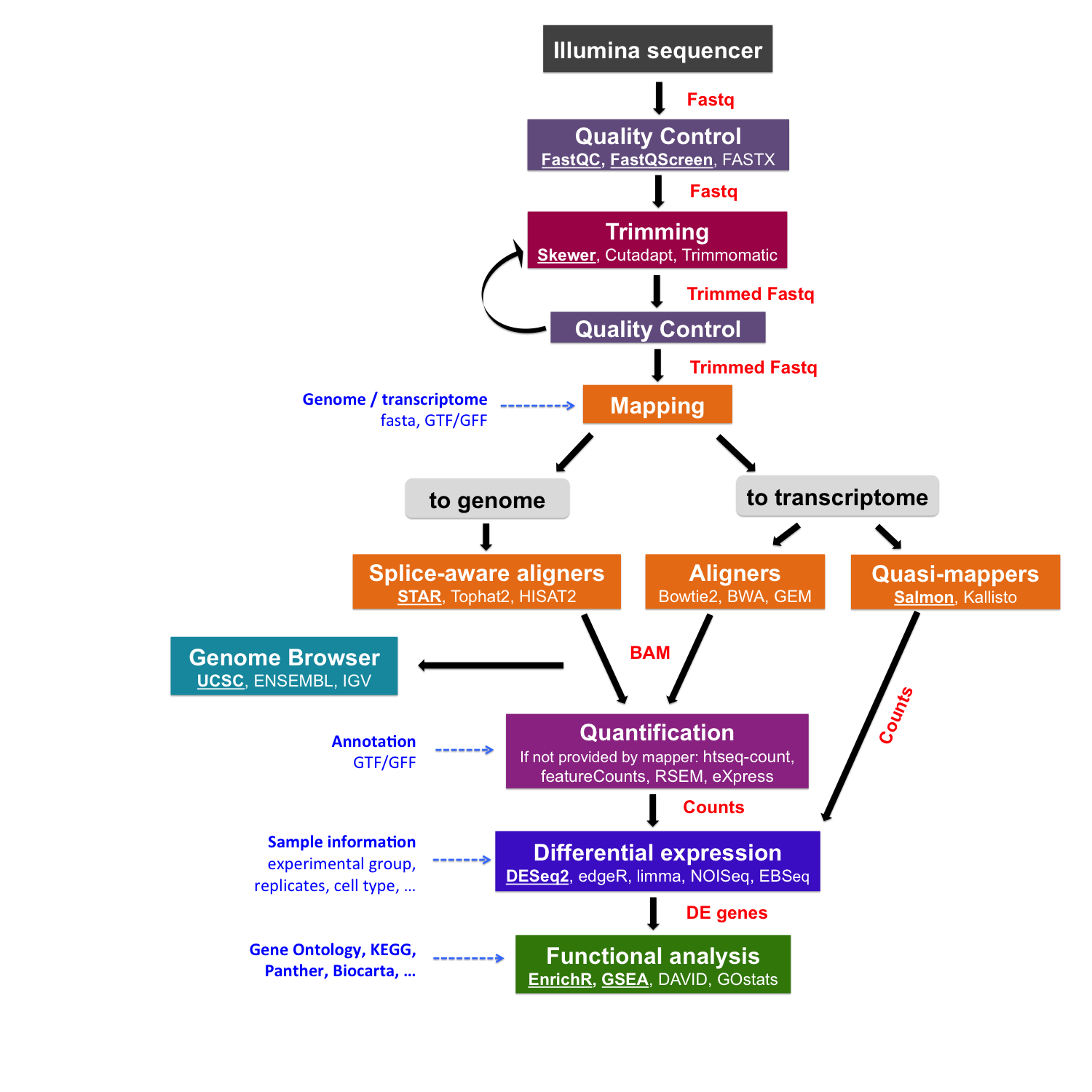
To appraise the quality of sequencing data, we will use the programs FastQC and Fastq Screen.
FastQC calculates statistics about the limerick and quality of raw sequences, while Fastq Screen looks for possible contaminations.
# Go to the "quality_control" binder cd ~/rnaseq_course/quality_control # Run FastQC for one sample $RUN fastqc ~/rnaseq_course/raw_data/fastq_chr6/SRR3091420_1_chr6.fastq.gz -o . # Run for all samples $RUN fastqc ~/rnaseq_course/raw_data/fastq_chr6/*fastq.gz -o . The output files are a .zip archive and an .html file
We can display the results (.html file) with an Net browser; e.m. Firefox:
firefox SRR3091420_1_chr6_fastqc.html & 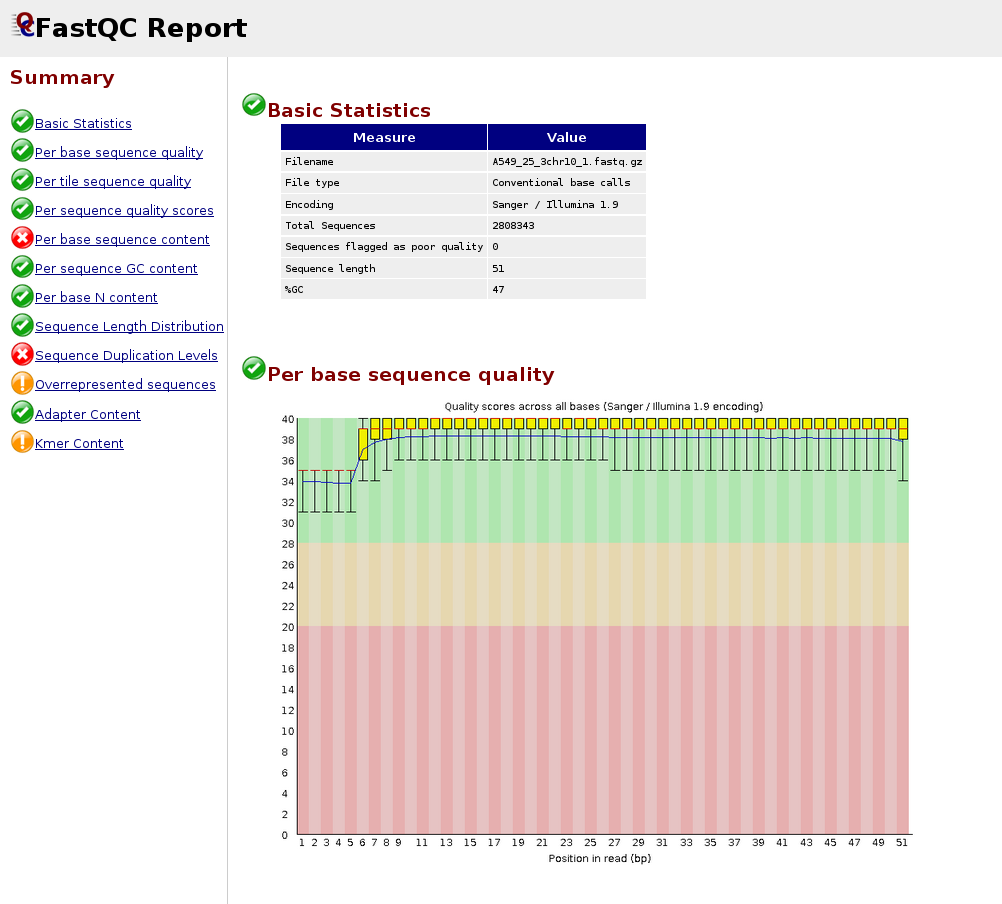
Beneath is an instance of a poor quality dataset. As you can see, the average quality drops dramatically towards the 3'-stop.
Yous can excerpt the files from the .null archive:
# extract unzip SRR3091420_1_chr6_fastqc.zip # remove remaining .naught file rm SRR3091420_1_chr6_fastqc.zip # display content of directory ls SRR3091420_1_chr6_fastqc File fastqc_data.txt contains the results in text format, for easier parsing of the results:
less SRR3091420_1_chr6_fastqc/fastqc_data.txt FastQ Screen
FastQ Screen is a tool that allows to screen libraries for potential contaminations.
It requires to download genome indices (data bases) from a variety of organisms, a lot of which tin can be downloaded by default.
Note that fastq_screen can exist establish in the singularity image, simply not the data bases!
WARNING: exercise not run the following command in grade: information technology will have too much time and resource!
# Download default data bases $RUN fastq_screen --get_genomes The latter commands download 14 indexes from Bowtie2 mapper (from model organisms or known contaminants):
- Arabidopsis thaliana
- Drosophila melanogaster
- Escherichia coli
- Man sapiens
- Mus musculus
- Rattus norvegicus
- Caenorhabditis elegans
- Saccharomyces cerevisiae
- Lambda
- Mitochondria
- PhiX
- Adapters
- Vectors
- rRNA
Upon download, the FastQ_Screen_Genomes binder is created, containing all indexes.
The file fastq_screen.conf will be also downloaded in this folder: in order to apply the tool, you will have to modify the fastq_screen.conf by providing the full path to the Bowtie2 executable (/usr/local/bin/bowtie2 if y'all use our singularity image) and total paths to the downloaded folders with genome alphabetize files.
Here we show where to change the executables:
# This is a configuration file for fastq_screen ########### ## Bowtie # ########### ## If the bowtie binary is not in your PATH and then you can ## ready this value to tell the plan where to find it. ## Uncomment the line beneath and prepare the appropriate location ## #BOWTIE /usr/local/bin/bowtie/bowtie BOWTIE2 /usr/local/bin/bowtie2 ... FastQ Screen runs checks on a random subset of 100,000 reads (that can be inverse using pick –subset).
You lot tin can execute FastQ Screen this mode:
$RUN fastq_screen --conf FastQ_Screen_Genomes/fastq_screen.conf \ ~/rnaseq_course/raw_data/SRR3091420_1.fastq.gz \ --outdir ~/rnaseq_course/quality_control/ Below is an example of the FastQ Screen results for SRR3091420_1.fastq.gz.
cd ~/rnaseq_course/quality_control/ # get files from fastq_screen run wget https://public-docs.crg.es/biocore/projects/training/PHINDaccess2020/SRR3091420_1_fastq_screen.tar.gz # extract tar -zvxf SRR3091420_1_fastq_screen.tar.gz firefox SRR3091420_1_screen.html 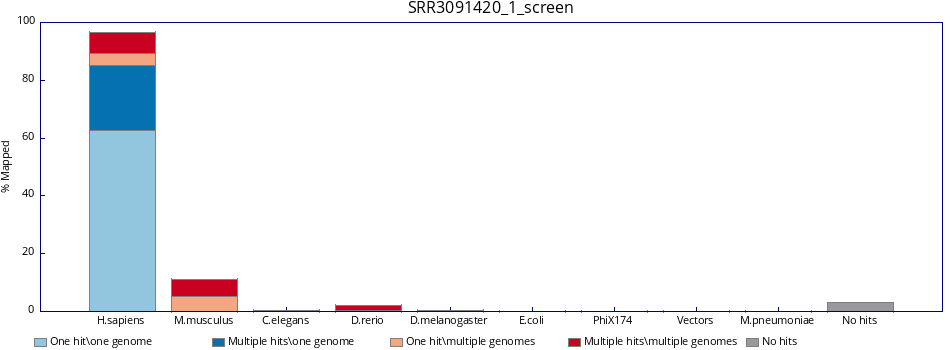
Initial processing of sequencing reads
Earlier mapping reads to the genome/transcriptome or performing a de novo associates, the reads have to be pre-processed, if needed, as follows:
- Demultiplex by index or barcode (usually washed in the sequencing facility)
- Remove/Trim adapter sequences
- Trim reads by quality
- Discard reads by quality/ambivalence
- Filter reads by thou-mer coverage (recommended for the de novo assembly)
- Normalize 1000-mer coverage (recommended for the de novo associates)
Every bit shown before, both the presence of low quality reads and adapters are reported in the fastqc output.
Adapters are unremarkably expected in small RNA-Seq because the molecules are typically shorter than the reads, and that makes an adapter to be present at 3'-end.
Below is an case of the FastQC report for a small RNA-seq sample:
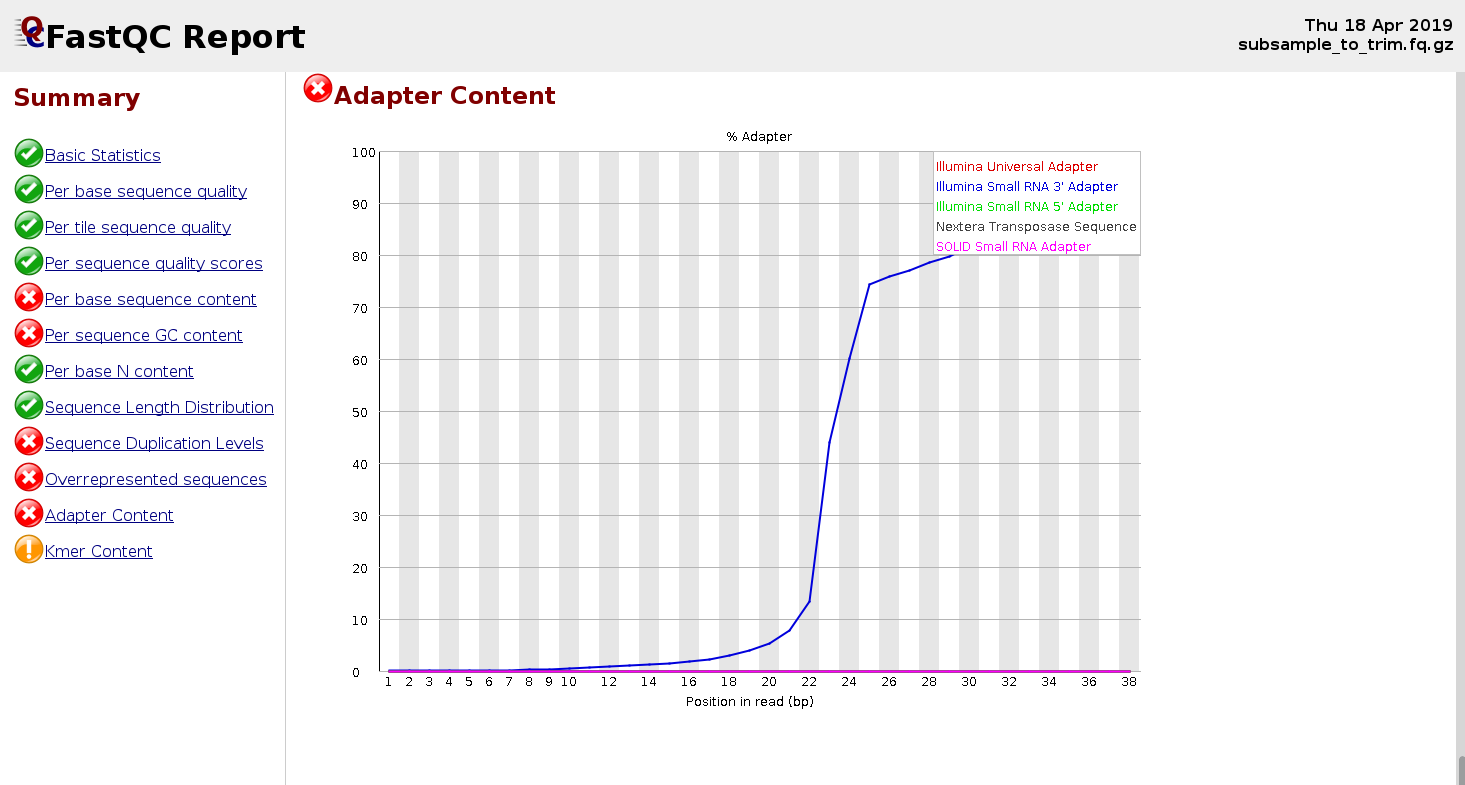
Trimming
There are many tools for trimming reads and removing adapters, such as Trim Galore!, Trimmomatic, Cutadapt, skewer, AlienTrimmer, BBDuk, and the most recent SOAPnuke and fastp.
Let'southward utilise skewer to trim the Illumina iii' adapter.
cd ~/rnaseq_course/trimming $RUN skewer ~/rnaseq_course/raw_data/fastq_chr6/SRR3091420_1_chr6.fastq.gz \ -x TGGAATTCTCGGGTGCCAAGG \ -o SRR3091420_1_chr6 \ -z .--. .-. : .--': :.-. `. `. : `'.' .--. .-..-..-. .--. .--. _`, :: . `.' '_.': `; `; :' '_.': ..' `.__.':_;:_;`.__.'`.__.__.'`.__.':_; skewer v0.2.2 [April iv, 2016] Parameters used: -- 3' end adapter sequence (-ten): TGGAATTCTCGGGTGCCAAGG -- maximum fault ratio allowed (-r): 0.100 -- maximum indel error ratio allowed (-d): 0.030 -- minimum read length allowed subsequently trimming (-l): 18 -- file format (-f): Solexa/Illumina 1.3+/Illumina 1.v+ FASTQ (automobile detected) -- minimum overlap length for adapter detection (-k): 3 Tue Feb 4 11:00:56 2020 >> started |=================================================>| (100.00%) Tue Feb 4 11:01:xl 2020 >> washed (43.669s) 12157169 reads processed; of these: 0 ( 0.00%) brusk reads filtered out subsequently trimming by size control 0 ( 0.00%) empty reads filtered out after trimming past size control 12157169 (100.00%) reads bachelor; of these: 480367 ( 3.95%) trimmed reads available afterward processing 11676802 (96.05%) untrimmed reads bachelor after processing log has been saved to "SRR3091420_1-trimmed.log". We can look at the read distribution after the trimming of the adapter by inspecting the log-file or re-launching FastQC.
cd ~/rnaseq_course/quality_control/ $RUN fastqc ~/rnaseq_course/trimming/SRR3091420_1_chr6-trimmed.fastq.gz -o . firefox SRR3091420_1_chr6-trimmed_fastqc.html & 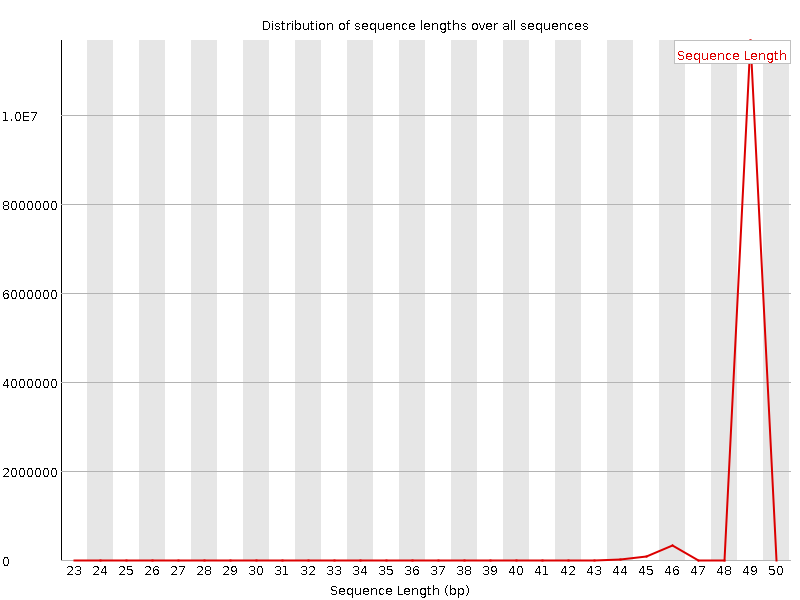
Case of a FastQC report for a trimmed modest-RNA sample:
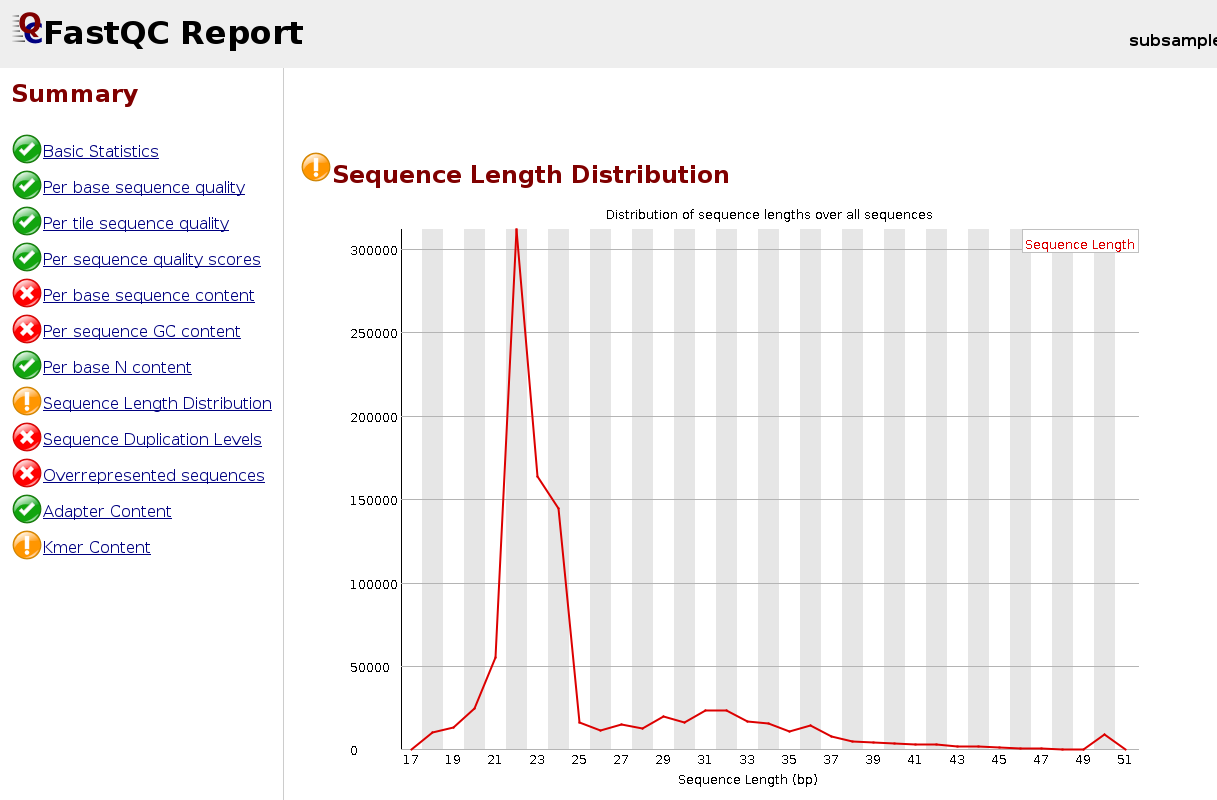
Now run skewer for all samples
for fastq in ~/rnaseq_course/raw_data/fastq_chr6/*fastq.gz do repeat $fastq $RUN skewer $fastq \ -ten TGGAATTCTCGGGTGCCAAGG \ -o $(basename $fastq .fastq.gz) \ -z done Do
Allow's explore the tool skewer in more detail: apply "skewer –help" for a description of the parameters.
- Which parameter indicates the minimum read length immune after trimming? What is its default value?
- Which parameter indicates the threshold on the average read quality to be filtered out?
- Using skewer, filter out reads in "SRR3091420_1-trimmed.fastq" that have average quality below 30 and trim them on iii'-end until the base quality has reached xxx. How many reads were filtered out and how many remain?
brownlithervard41.blogspot.com
Source: https://biocorecrg.github.io/PHINDaccess_RNAseq_2020/qc_trimming.html
{kind=link}
Post a Comment for "Trim Galore Not Removing 0 Length Reads"